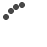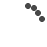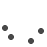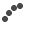Thrombotic Thrombocytopenic Purpura (TTP) is a rare but critical TMA( Thrombotic Micro Angiopathies) caused by severe deficiency of the ADAMTS13 metalloprotease, typically due to auto-antibodies. This deficiency leads to ultra-large von Willebrand factor (vWF) multimers accumulating, causing uncontrolled platelet aggregation and microthrombi formation, resulting in MAHA, thrombocytopenia, and organ ischemia.
TTP is characterized by MAHA with severe thrombocytopenia and variable organ ischemia, most commonly neurologic, cardiac, or renal. The diagnosis is confirmed by a severe deficiency (<10%) of ADAMTS13 activity. TTP is further divided into two categories based on the mechanism of ADAMTS13 deficiency: congenital (inherited) vs. immune-mediated (acquired). Congenital TTP, also known as Upshaw–Schulman syndrome or hereditary TTP, is defined by a persistent severe deficiency (<10%) in ADAMTS13 caused by biallelic pathogenic mutations in the ADAMTS13 gene. Immune-mediated TTP, sometimes referred to as acquired TTP, is caused by ADAMTS13 deficiency mediated by auto-antibodies. iTTP is further subdivided into primary iTTP, when there is no obvious associated disorder, and secondary iTTP, when an associated condition can be identified.
Mrs. X, 46-year-old female, no known comorbidities. Presented with history of fever with headache for one day. On examination- Patient was conscious, oriented, febrile. Pallor and mild Scleral Icterus noted. No neck stiffness. Blood Pressure: 120/80mmHg.PR- 98/min. Systermic examination was unremarkable.
Lab investigations were done which showed Hemoglobin (Hb): 8.5 g/dL,Total WBC Count: 5870/ mm3, Platelet Count: 28,000 /mm3 (Severe Thrombocytopenia), MCV: 86.4/ fL, Creatinine: 0.64 mg/dL, Urea: 16.3 mg/dL, Total Bilirubin- 1.69 mg/dl, Direct Bilirubin– 0.53 mg/dl, Indirect Bilirubin – 1.16 mg/dl, Liver enzymes Normal. USG Abdomen: Showed Cholelithiasis without features of cholecystitis.
Work up for fever was done. Dengue NS1 and serology, scrub typhus IgM, Malarial parasite negative. Peripheral Smear reports showed Normocytic normochromic with few RBC fragments and many polychromatic RBCs and Moderate thrombocytopenia, with an impression of Bicytopenia. In view of the presence of schistocytes (RBC fragments) with associated anemia and thrombocytopenia, further workup was suggested to rule out a microangiopathic hemolytic anemia (MAHA). As suggested, the hemolysis workup was done revealing LDH- 655.1, Direct coombs test positive, C3 – 85.26 (low), C4 – <4 (low), Reticulocyte – 8.75 % (High)- suggestive of an ongoing hemolysis.
Following which an autoimmune workup was done. ANA Profile – SS-A: Borderline (+) and SS-B: Strong (++), which was suggestive of an underlying autoimmune etiology. P and C ANCA Negative. Beta 2 glycoprotein IgM and IgG- negative. Anticardiolipin antibody IgM and IgG – negative.
Patient had persistent headache which didn’t resolve with analgesics, in view of which neuroimaging was done. MRI Brain showed tiny diffusion restricting T2W/FLAIR hyperintense focus in the right parietal region (possibility of acute lacunar infarct). MR Angiogram/Venogram: No evidence of stenosis, occlusion, or cerebral venous thrombosis.
Initial Concern was anemia and thrombocytopenia with hemolysis , highly suggestive of an autoimmune process. Hematologist was consulted for the same . Rheumatologist was consulted due to positive ANA findings (SS-B, SS-A). Neurologist was consulted regarding the MRI findings and advised to continue conservative management with observation. Patient was conservatively managed. Platelet counts improved. Patient was clinically and hemodynamically stable at the time of discharge.
Approximately six weeks later, patient presented with abdominal pain for three days associated with nausea and one episode of vomiting. History of loose stools for 1 day. Generalised tiredness/weakness. Headache with transient slurring of speech on and off . No history of frank bleeding, hematuria, dysuria, chest pain, or shortness of breath. The initial clinical impression with this presentation was concerning for a hemolytic/thrombocytopenic crisis or exacerbation of her known autoimmune condition. Patient had recurrent episodes of transient slurring of speech.
The workup was immediately tailored to rule out TTP and other secondary causes of MAHA/Thrombocytopenia. Blood tests demonstrated a severe MAHA flare- Hemoglobin has dropped from 7.7 g/dL to 5.9 g/dL- following which 1 pint PRBC Transfusion was done, Platelet Count – Severe thrombocytopenia, dropping from 29,000/mm3 to 5,000/mm3 – 4 RDP transfused, LDH- 1266.1 U/L, Bilirubin (Total): Elevated and rising, peaking at 6.14mg/dL. Peripheral Smear – similar picture to previous admission- schistocytes- suggestive of MAHA. Reticulocyte count – 22.6 %, Complement C3/C4 – low, direct coombs test- positive
Image of peripheral smear showing schistocytes
ADAMTS13 level- 0.01 IU/ml ( low ) and Anti-ADAMTS13 inhibitor Antibody – Negative
Hematologist and Rheumatologist were consulted. Bone Marrow Aspiration and Biopsy (BMAB): A BMAB was planned and performed under IV sedation to rule out other possible causes of bicytopenia and bone marrow involvement. Steroid Pulse Therapy was initiated based on the high suspicion of an autoimmune process. Day 1: 500 mg IV Methylprednisolone over 4 hours. Day 2 & 3: High dose Methylprednisolone 1000 mg IV over 4 hours.
In view of persistent drop in platelets, A plan was established to initiate Steroid Pulse + PLEX (Therapeutic Plasma Exchange), the critical first-line therapy for suspected TTP. Two cycles of PLEX were done on alternate days. CBC was repeated daily to track response to therapy, specifically the platelet count and hemoglobin level.
Patient’s platelets improved drastically and reached 4 lakhs post two cycles of plasma exchange and planned for discharge. Continued on steroids Inj Dexamethasone 8mg IV OD and then tapered and converted to oral steroids. Since patient showed a satisfactory response to plasma exchange, no further rituximab was planned.
This case presents a compelling diagnostic and therapeutic challenge involving a 46-year-old female with a strong prior history of autoimmune hemolytic disease who presents with an acute, life-threatening Thrombotic Microangiopathy (TMA) flare.
The differential Diagnosis were TTP vs. Autoimmune Hemolytic anemia with thrombocytopenia. The core diagnostic dilemma lies between an acute episode of Acquired Thrombotic Thrombocytopenic Purpura (aTTP) and an exacerbation of AIHA particularly as the patient has a known history consistent with the later.The combination of severe, unexplained microangiopathic hemolytic anemia (MAHA), severe thrombocytopenia and neurological symptoms strongly raises the clinical suspicion for TTP. While AIHA is a possibility, the clear signs of MAHA (high LDH, schistocytes) necessitate immediate TTP protocol initiation. Decreased ADAM-TS 13 levels and activity pointed towards TTP.
TTP is a hematologic emergency with an estimated mortality rate exceeding 90% if untreated. Therefore, the decision to initiate TTP-directed therapy based on clinical and laboratory suspicion, without waiting for the definitive ADAMTS13 result, is correct and life-saving.First-Line Therapy: Therapeutic Plasma Exchange (PLEX) combined with high-dose corticosteroids (Methyl prednisolone pulse) is the standard-of-care for suspected TTP. PLEX rapidly removes the offending autoantibodies (anti-ADAMTS13) and replenishes functional ADAMTS13 enzyme. Steroids: The use of high-dose steroids provides rapid immune suppression, targeting the underlying autoimmune production of the anti-ADAMTS13 antibodies (and managing the underlying Evans/Autoimmune component).
The Plasmic score helps to decide on starting plasma exchange in a suspected TTP
score
transplant
0–4- Low (<5%)- TTP unlikely
5 Intermediate (~50%)- Possible TTP
6–7 High (>90%) -TTP very likely -Start
plasma exchange
A score of 7 in our case – which indicates TTP is highly likely
The role of definitive testing and targeted therapy is important.The ADAMTS13 activity assay (deficiency defined as activity <10%) is crucial to confirm the diagnosis of TTP. Rituximab and Caplacizumab can be considered , given the patient’s relapsing course and established autoimmune history. Rituximab (an anti-CD20 monoclonal antibody) is often used as a second-line or prophylactic agent in TTP to suppress B-cell activity and prevent relapse by decreasing autoantibody production. Caplacizumab (a Nanobody) is a potent, modern adjunct therapy. It works by binding to the A1 domain of von Willebrand factor (vWF), physically blocking the interaction between the ultra-large vWF multimers and platelets. This rapidly halts microthrombi formation and protects the patient from ischemic organ damage and rapid platelet consumption while waiting for PLEX and steroids to address the underlying immune deficiency.
The distinction is crucial. Autoimmune Bicytopenia is managed with immunosuppression (e.g., steroids, rituximab), while TTP requires immediate PLEX. The presentation of a TMA in the context of known autoimmune disease necessitates rapid differentiation and aggressive treatment to prevent fatal outcomes.
The patient’s underlying autoimmune condition makes her susceptible to secondary TTP, or a “TTP-like” syndrome driven by severe endothelial damage. While the positive ANA profile points to a broader CTD/autoimmune context, the clinical presentation of the second flare meets the threshold for empirical treatment of TTP.
The appropriate initial management was the prompt initiation of PLEX and pulse steroids for suspected TTP. Future management will depend on the definitive ADAMTS13 results, and consideration of Rituximab or potentially Caplacizumab should be given if no response to the current therapy. Also, this case highlights the necessity of maintaining a high index of suspicion for TTP in any patient with underlying autoimmune hematologic disorders who develops new MAHA features, even with atypical presentations.
Inhibitor antibodies were negative. Though isolated SSB was strong positive, the patient does not clinically fit into any autoimmune condition at present.The entire sequence of events were triggered by infection
Serology revealed a positive ANA with specificities (SS-A borderline, SS-B strong positive), highly suggestive of an underlying Connective Tissue Disorder. Direct Coombs Test was positive.
Finally, placing the diagnosis predominantly in the realm of primary TTP. However, the patient’s strong underlying autoimmune background, including a prior positive ANA profile (SS-A, SS-B), necessitates cautious long-term follow-up for a relapsing autoimmune condition. Given the overlap, the TTP may be better classified as secondary TTP related to an undifferentiated connective tissue disease (CTD), or the patient may have two distinct autoimmune processes. Therefore, future surveillance must be vigilant for TTP relapse (guided by ADAMTS13 levels monitoring) and the possible evolution into a definite systemic autoimmune disease that could trigger future secondary TTP or other secondary TMA events.
Dr N Roseline Sweety Jebapriya DNB First year Resident General Medicine, Kauvery Hospital, Chennai.
Dr S. Sivaram Kannan Clinical lead and Chief consultant Physician, Kauvery Hospital, Chennai.
Dr Arshad Raja Consultant Hematologist, Kauvery Hospital, Chennai.
Dr Sham Consultant Rheumatologist, Kauvery Hospital, Chennai.