Amyotrophic Lateral Sclerosis (ALS): A case report
Anuradha. S
Clinical Pharmacist, Kauvery Hospital, Hosur, Tamil Nadu
Introduction
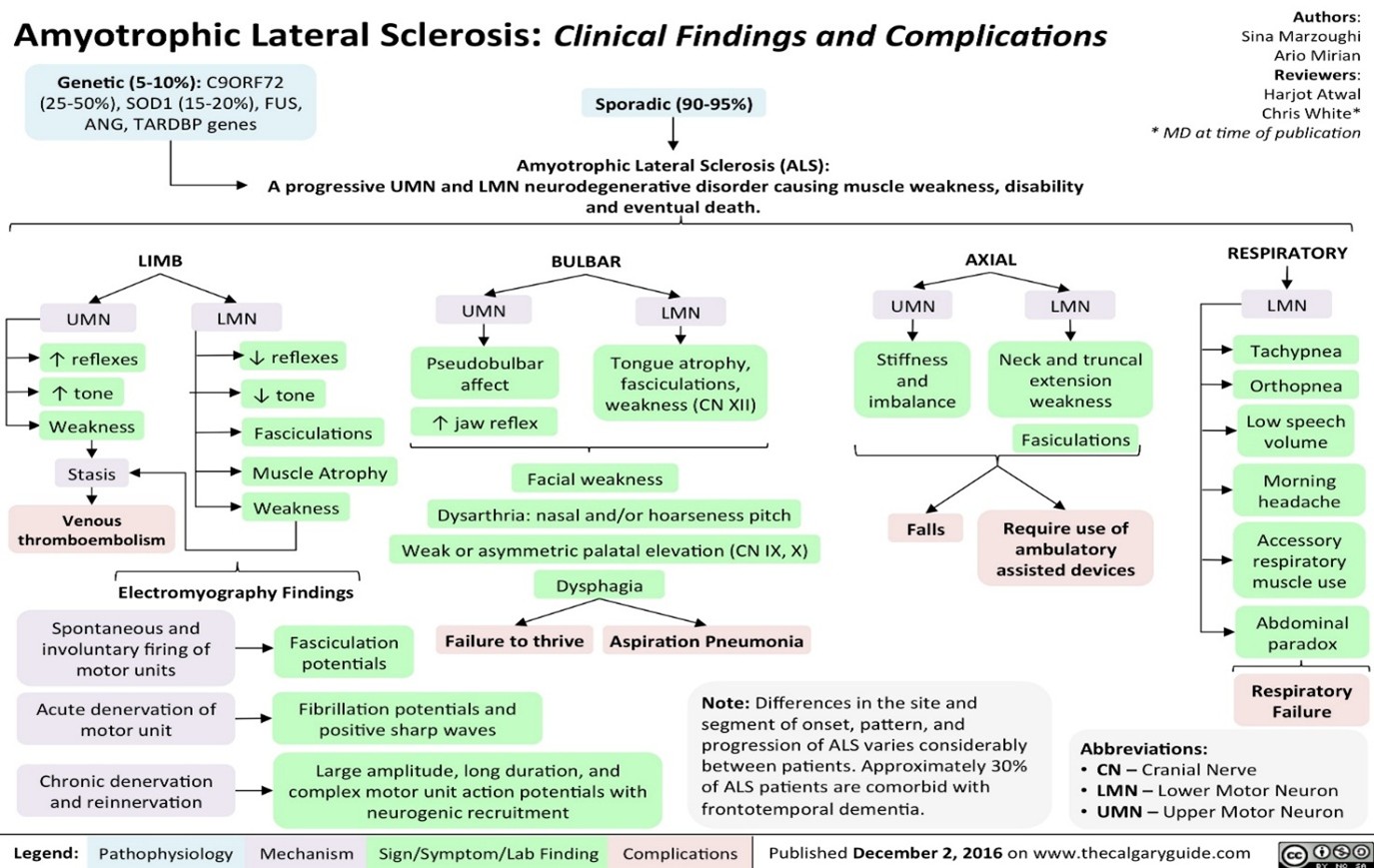
Amyotrophic lateral sclerosis (ALS), is a nervous system disease that affects nerve cells in the brain and spinal cord. ALS causes loss of muscle control. ALS is often called lou gehrig’s disease. It is characterised by rapidly worsening muscular paralysis brought by motor neurone degeneration, which results in muscle atrophy, spasticity, and vulnerability. The exact cause of the disease is still not known. A small number of cases are inherited. ALS often begins with muscle twitching and weakness in an arm or leg, trouble swallowing or slurred speech. Eventually ALS affects control of the muscles needed to move, speak, eat and breathe. There is no cure for this fatal disease. Amyotrophic lateral sclerosis can manifest clinically in a variety of ways. The diagnosis is crucial for both the prognosis and the course of treatment. ALS is typically indicated by both upper motor neurone and lower motor neurone sings as well as electrodiagnostic tests.
Clinical manifestations on ALS
Symptoms
ALS vary from person to person. Symptoms depend on which nerve cells are affected. ALS generally begins with muscle weakness that spreads and gets worse over time. Symptoms might include: trouble walking or doing usual daily activities, muscle twitching, tripping and falling, weakness in the legs, feet or ankles, hand weakness or clumsiness, slurred speech or trouble swallowing, weakness associated with muscle cramps and twitching in the arms, shoulders and tongue, untimely crying, laughing or yawning, thinking or behavioral changes.
Cause
It also usually doesn’t affect the senses, including the ability to taste, smell, touch and hear.ALS affects the nerve cells that control voluntary muscle movements. These nerve cells are called motor neurons.
There are two groups of motor neurons:
- The first group extends from the brain to the brain stem and spinal cord. They’re referred to as upper motor neurons.
- The second group extends from the brain stem and spinal cord to muscles throughout the body. They’re referred to as lower motor neurons. ALS causes both groups of motor neurons to gradually deteriorate and then die.
Complications on ALS
As the disease progresses, ALS causes complications, such as:
- Breathing problems, speaking problems, eating problems, dementia
- The most common cause of death for people with ALS is breathing failure. Half of people with ALS die within 14 to 18 months of diagnosis. However, some people with ALS live 10 years or longer.
- Some people with ALS have problems with language and decision-making.
- Some are eventually diagnosed with a form of dementia called frontotemporal dementia.
Case Presentation
Patient Mr. 44y/male, came to opd with above mentioned complaint and shifted to ward for further management. he was evaluated and found to have Amyotrophic lateral sclerosis. strict vitALS & i/o chart were monitored. patient was managed with iv Inj.edaravone, Inj.methylprednisolone and other supportive measures. patient symptomatically better and hence discharged with following advice.
Subjective
Current Complaints (Cc)
- Patient came to opd with c/o difficulty using both upper limbs: 1 years
- C/o walking difficulty: 2-3 months’ progressive
- Started as difficulty holding object with left hand which progressed to difficulty raising left arm followed by right upper limb. Over past 6 months, difficulty in buttoning. Over 3 months, couldn’t mix and eat food with right hand.
- H/o twitching of left arm muscles
- H/o / difficulty raising left leg especially on getting up from sitting. Needs to hold railings to climb stairs
- Feels left leg stiff
- Past Medical History: No Comorbidities
- Past Medication History: Nil
On Examination
- Conscious, Oriented
- Cranial Nerves – Normal Except Tongue Fibrillations
- Muscle Fasciculations Present
- All Dtrs Brisk/Exaggeratted Polyminimyoclonus Was Present
- Touch, Pain, Vibration – Normal
- Power – Small Muscles of Hand Weak
- Elbow Grade 4
- Shoulders Grade 3 To 4
- Proximal Lower Limbs Grade 4 – On Left
- Grade 4 On Right
- Ankle Clonus (+) Right
- Plantar – Extensor On Right
Objective
| Physical Examination | |
|---|---|
| BP | 130/80 mmhg |
| Pulse rate | 98/Min |
| Respiratory rate | 22/Min |
| Temperature | 97.20f |
| Spo2 | 97% On Room Air |
| Other Systemic Examination | Normal |
Vital Signs
| Vital/Date | Day 1 | Day 2 |
|---|---|---|
| Temp (F) | 97.20f | 97.20f |
| Pulse /Min | 98/Min | 98/Min |
| Bp (Mm/Hg) | 130/80 Mmhg | 130/80 Mmhg |
Laboratory Investigation
Department of Hematology
| Constituents | Status | Detected Values | Normal Values | Unit |
|---|---|---|---|---|
| WBC Count | Normal | 7710 | 4000 - 10000 | Cells/Cu Mm |
| Monocytes | High | 9.7 | 1-8 | % |
Department of Biochemistry
| Constituents | Status | Detected Values | Normal Values | Unit |
|---|---|---|---|---|
| Potassium | Normal | 3.6 | 3.5-5 | Mmol/L |
| Urea Serum | Normal | 12.8 | 10 -50 | Mg/Dl |
| Sodium | Normal | 139 | 136-145 | Mmol/L |
| Creatinine | Normal | 0.6 | 0.5-1.4 | Mg/Dl |
Drug-Drug Intractions: Nil
Advice on discharge
| Drug | Dose | Frequency | Duration | Af/Bf |
|---|---|---|---|---|
| Cap.Riluzole | 50 Mg | BD | 1 Month | After Food |
| Injection. Edaravone 60mg As Infusion Over 1 Hour Od X 12 Days | ||||
Assessment of therapeutic response of Edaravone and Riluzole combination therapy in Amyotrophic Lateral Sclerosis patients.
Discussion
Edaravone and Riluzole are both FDA-approved ALS drugs, but they work differently: Riluzole extends survival slightly by reducing glutamate, while Edaravone slows functional decline by fighting oxidative stress, particularly effective early on. Combination therapy, often using both, shows promise, with Edaravone potentially offering better functional maintenance using both drugs targets different pathways (glutamate & oxidative stress) for potentially greater benefit gives synergistic effect. Efficacy of Edaravone provides better functional preservation (ALSfrs-r) when used with riluzole compared to riluzole alone. The association between riluzole and edaravone with liver damage and gastrointestinal discomfort underscores the need for enhanced safety monitoring in patients with hepatic dysfunction. Despite these side effects, riluzole, edaravone, remain essential in ALS management. These medications can slow disease progression and extend survival, making them invaluable in the absence of a cure. However, clinical practice must prioritize personalized treatment approaches that balance therapeutic efficacy with potential risks to ensure patient safety and quality of life. Future research should focus on the long-term impact of these side effects and strengthen drug safety monitoring to optimize treatment strategies and advance personalized ALS therapies.
Conclusion
Combined with riluzole, edaravone slows disease progression and is safe, but the effect is short-term. Bulbar symptoms respond better to combination therapy. The serum creatinine is helpful in monitoring disease progression. Survival rate of real-world studies show lower mortality risk with edaravone (often combined with riluzole). Offers broader protection, with edaravone potentially enhancing riluzole’s effects, though safety monitoring remains important.
Nightingale Journals
- Editorial
- Nursing care of patient with arrhythmia in various scenario at non critical and critical ward
- Arrhythmia Identification and Differentiation
- Pediatric arrhythmias diagnosis and management in children
- Understanding electrocardiography fundamentals and its interpretation
- Non-Pharmacological management of arrhythmias
- The science behind arrhythmias: Pathophysiology and causes
- A comprehensive review on ABG Analysis
- Airway Management: An overview
- Exploring different ways to deliver oxygen
- Management of raised ICP
- Paediatric early warning score: Good tool for clinical practice
- Enhancing continuity and safety in critical care: Through effective documentation and structured handover
- Implementation of evidence-based care bundles to prevent healthcare-associated infections
- Prevention and management of pressure injuries in clinical settings
- Safe transport of critically ill patients: Principles, protocols and best practices
- The vital role of nurses in critical care procedures: A focus on pre-procedure preparation
- Anatomy and Physiology of Kidney
- Anemia in Kidney Disease
- Fluid and Electrolyte Imbalance
- Hypertension and diabetes mellitus management in kidney disease
- ESRD: Palliative Care
- Care of Hemodialysis and peritoneal dialysis patients
- In house Continuing Nursing Education (CNE) on “Diabetic Foot Care” at Kauvery Hospital, Cantonment
- Patient Education on Foot Care Nursing Role
- In-House-Continuing Nursing Education (CNE) on “Basic Concepts of Nursing in Polytrauma” Kauvery Hospital, Tennur – 2025
- Assessment and Resuscitation of Head Injury
- Initial Assessment of Trauma Patients
- Monitoring and Prevention of complications During the Transfer of Polytrauma Patients
- Role of Critical Care Nursing in Polytrauma Management
- Anatomy and Physiology of Skin
- Diabetic Wound Care Management
- High Risk for Developing Pressure Injuries
- Medical Device Related Pressure Injury
- Prevention of Surgical Site Infections
- Wound care: An overview
- Wound Healing: An overview
- Editorial
- A case of hypoxic ischemic encephalopathy
- A case of multiple tablets overdose
- From elective surgery to mechanical ventilation: A critical post sterilization nursing care: A case report
- A rare case of Kikuchi disease with recurrent symptoms
- A journey from thrombolysis to decompressive craniectomy and lifesaving fasciotomy
- Arrhythmogenic right ventricular dysplasia presenting as ventricular tachycardia in an adolescent: A case report
- Deep right capsuloganglionic high-flow arteriovenous malformation presenting with intracerebral hemorrhage in an adolescent: A comprehensive case report
- Posterolateral thoracotomy for tumor excision
- Ventricular septal defect repair
- Cardiac myxoma and beyond: A case of benign tumor
- Learned Helplessness in Maternal Health: Implication for Maternal Well-being and the Transformative Role of Nurses
- Allergic Bronchopulmonary Aspergillosis (ABPA): The medical nutrition therapy for ABPA
- Handgrip dynamometry and anthropometric measurements for assessing nutritional repletion in malnourished cancer patients undergoing nutrition support
- Sudden cardiac arrest with super-refractory seizures in a patient with bilateral temporal lobe meningioma and end-stage renal disease
- Spectrum of supraventricular and ventricular arrhythmia’s: A case series highlighting AVNRT and arrhythmogenic right ventricular dysplasia
- The combination approach of rosuvastatin and ezetimibe in post-stent patients
- Hypoglycemic encephalopathy with diffuse cerebral edema and multi‑organ dysfunction: Perspective on a patient with type 2 diabetes mellitus
- Complicated UTI with MDR Pseudomonas in an elderly patient managed with combination antibiotics and DJ stenting
- Non-Obstructive Hypertrophic Cardiomyopathy presenting with asymptomatic: A Case Report
- Awake craniotomy for left frontal high-grade glioma: a multidisciplinary case report
- Journal Scan: A review of images in clinical medicine of immediate clinical significance, harvested from major international journals
- Editorial
- A case of Acute C5 – C6 Compressive myelopathy with bowel involvement
- A Case of Acute lymphoplastic leukemia
- A case of cerebellar hemangioblastoma causing obstructive hydrocephalus
- A case of thymoma
- Acute food-induced anaphylaxis in an elderly male with comorbidities
- A case study on congenital cyanotic heart disease with Ventricular Septal Defect (VSD) managed by surgical closure with RVOT resection
- Calcific AS and TAVI
- Conduction System Pacing (CSP)
- A case report on DCLD with hepatic encephalopathy and multidrug resistant sepsis
- Surgical management of necrotizing pneumonia with advanced empyema in a 3-year-old child: A case report
- Comprehensive approach to tuberculosis care: insights from a regional workshop at Kauvery hospital, Electronic city
- Hypokalaemia induced paralytic ileus
- Role of bubble contrast echocardiography in the diagnosis of hepatopulmonary syndrome
- Cerebral venous thrombosis with intracerebral hemorrhage in a patient with C1Q nephropathy and acute pancreatitis: a rare clinical constellation
- A retrospective study to assess postoperative pain levels among patients undergoing IPOM and E TEP hernia repair
- Bilateral uterine artery embolization
- Paraquat Poisoning
- From clinical examination to CT diagnosis: An emergency nurse–led approach to acute abdominal pain
- Unmasking hidden hypoxia: A case of methemoglobinemia in a young adult
- Emergency nursing management of massive upper GI bleed
- Successful management of severe sepsis with respiratory failure following live renal transplantation
- Acute Fatty Liver of Pregnancy
- Tracheoesophageal fistula repair
- Message from Dr. Manivannan Selvaraj, Founder and Managing Director, Kauvery Hospitals
- EDITORIAL
- Editorial Board
- Voices from the field!
- Junior nurses in Kauvery Hospital on the frontline against the COVID-19 pandemic
- Challenges in nursing care in achieving a successful outcome to a Liver Transplant on a 2-year old child with life threatening genetic condition
- Nursing management of a multi-organ transplant (Liver and Kidney) on an adolescent
- Successful discharge of a four year old baby with life threatening gas gangrene and compartment syndrome
- The revolutionary effect of a nursing-driven initiative to reduce ICU re-admissions
- Nephrotic syndrome: A case report
- Nursing Care of Patient with Thoracic Endo vascular Aortic Repair (TEVAR)
- Efficient Nursing Care equals Early Recovery
- Umbilical Cord Blood Banking is a lifesaver for your family, “Create one life, save another”: A review
- Challenges associated with wearing gloves
- Certificate Course on Infection Control 2022
- The ANEI Young Leader Award, 2022, comes to Kauvery Hospitals!
- The AHPI Nursing Excellence Award 2022, comes to Kauvery Hospital, Tennur
- Editorial
- Editorial Board
- Caring is the Essence of Nursing
- To be patient and calm! Why I Love Working as an Emergency Room Nurse
- Infective Endocarditis
- Infective Endocarditis
- Patent Ductus Arteriosus
- HOSPITalk 2023 – Clinical Governance
- Cardiac healthy diet
- Healthy diet for people working night shifts
- Chimeric antigen receptor (car) T cell therapy
- Human papillomavirus (HPV) vaccine
- Kauvery Hospital Journey on Winning National Level CII QC Competition Against the Industrial Sectors
- செவிலியர்கள் தின வாழ்த்து கவிதை
- அன்பு காவேரி
- Hepatology for nurses
- Role of Physiotherapy in Traumatic Brain Injury: A case report
- My experience at Kauvery Hospital, Salem
- Editorial
- Editorial Board
- Benign Paroxysmal Positional Vertigo (BPPV): A Case Series
- Nutrition for Nurses
- Cardiovascular effects of sodium – glucose co-transporter- 2 inhibitor, Glucagon-like Peptide-1 Receptor Agonists and Dipeptidyl-peptidase 4 inhibitors
- Brief Guide on Food Safety Standards to be Adopted Summer in South India
- Kauvery Hospital: Pioneering the Future of Healthcare with IoT, Wins Healthcare Asia Awards.
- மருத்துவர்கள் தின வாழ்த்து கவிதை
- காவேரியனின் தன்னம்பிக்கை
- வெற்றியின் பாதை
- Editorial
- Editorial Board
- A six-year-old girl gets a Permanent Pacemaker implanted!
- Cardio Renal Amyloidosis (Non-Secretory Myeloma)
- Deep Vein Thrombosis, treated with Angiojet Percutaneous Mechanical Thrombectomy (PMT): A Case Report
- Emergency CABG in Acute MI
- Meatotomy
- Septic Shock with Multiple Organ Dysfunction Syndrome: A Case Report
- Stab Injury: A Case Report
- The Journey of Dialysis Unit at Hosur
- The measure of life is not its duration but its donation
- Innovation at the frontline of Nursing: A Review
- A case study on hydatid cyst
- Untold History of British Scientist: Contributed to the Discovery of DNA
- Neurology for Nurses
- Poem
- மருத்துவாலயம்
- EDITORIAL
- Editorial Board
- Biological or circadian clock
- A novel direct thrombin inhibitor from tick salivary transcriptoms: A review
- Nursing Care of a Patient with Coarctation of aorta (COA)
- Nursing care of patient with aortic dissection
- Wireless moonlight: An ultra-short case study
- Acute liver failure: A case report
- We nurses hand-hold a patient on a long walk down a dark COVID road
- Effectiveness of segmental breathing and active cycle of breathing technique in the management of dyspnoea among covid-19 patients
- EDITORIAL
- Editorial Board
- I am a proud to be a Liver Transplant Nurse!
- Head Injury: A case report
- Postpartum/Peripartum Cardiomyopathy
- Nursing Care of Patient with Myocardial Infarction
- Nursing challenges faced in care of patient with Blunt Injury Abdomen
- Salicylate poisoning
- Vascular Access Management (VAM) training program for nurses
- Therapeutic Nutrition among Critical care patients
- Diagnostic Image: Palmar Hyperlinearity and Filaggrin Deficiency
- Journal Scan
- EDITORIAL
- Editorial Board
- Guideline-directed drug treatment for heart failure
- Dilated cardiomyopathy
- IV Proton Pump Inhibitors (PPIs): What are the true indications?
- Role of physiotherapy in GBS patients during hospitalization:A case presentation
- Pulmonary Thromboembolism
- Paraquat poisoning, an emerging problem, a challenging outcome
- Achievements of today are the stepping stones for success tomorrow!
- Diagnostic Image
- Chronic obstructive pulmonary disease: A case report
- EDITORIAL
- Editorial Board
- The Good Nurse
- Success story of a patient with Haemangioma
- Role of physiotherapy in Bell’s Palsy at outpatient department
- Arrhythmogenic Right Ventricular Dysplasia: A Cardiomyopathy
- Systemic Lupus Erythematosus (SLE)
- Teamwork makes a Dreamwork
- Cholecysto Cutabeous Fistula, an emerging problem: a challenging outcome
- Adhesive small bowel obstruction: Nutrition care process
- Diagnostic Image: Moderate Pitting Edema in Lower Limbs
- Editorial
- Editorial Board
- Quality improvement project to reduce the incidence of ventilator-associated pneumonia
- Rare disease, desired outcome
- Management of patients with acute myocardial infarction with ischemic stroke
- Left mediastinal tumor excision
- Esophageal varices grade III: A case report
- TB meningitis: A case report
- Nutrition care process for Sigmoid Diverticulitis
- Nutritional management of patient who underwent emergency laparotomy and GIST
- Nutritional management of gestational diabetes mellitus
- Nutrition and drug interaction
- மக்களின் நம்பிக்கை!
- காவேரித்தாய் – 2
- ஊசியின் மகத்துவம்
- Editorial
- Editorial Board
- Resilient and Empowered: The Unstoppable Force of Women’s Spirit
- “Global Perspectives on Patient Safety: A Recap of the International Patient Safety Conference – 13th to 14th Feb 2023”
- Acute Pancreatitis: the nutrition care process
- Role of diet in mitral valve replacement: A case presentation
- Our colleague’s SVT (Supra Ventricular Tachycardia)!
- Overview of Acute Respiratory Distress Syndrome
- To prevent the negative impact of Inj. Amphotericin among Mucormycosis patients
- Diagnostic Cases: Spontaneous Abortion and Gangrene
- Effective Adherence of Checklist through Digitalisation
- Nephrology for Nurses
- “உயிரைக்காக்கும் உயரியதானம்”
- மக்களின் நம்பிக்கை
- Nightingale Journal Editorial – April Issue Highlights
- Editorial Board
- SVT (Supra Ventricular Tachycardia)
- Tender loving care of patient with DVT after IVF (in vitro fertilization)
- ASD Surgical Closure
- Quality improvement project to reduce the risk of cross infection from wall mounted suction apparatus
- Quality Improvement Project on Crash Cart Management with numbered seal
- Log the Indian Millets
- Crystal your body by dint of seven Crystal Seeds!
- Role of Glucagon
- Educative Image: Department of Orthopaedics
- Nephrology for Nurses! – Part 2
- EDITORIAL
- Editorial Board
- இதயநகரத்தின் செவிலியர்கள் தின வாழ்த்து
- My experience as a patient
- A study on Anidulafungin therapy in oncology patients in a tertiary care hospitals
- A case study on Heart Transplantation
- Your Physiotherapist’s talks! Know your Moves
- Impact of air pollution on human health
- 4th QOK Group Level Competition 2023
- 7th International conference of CAHO
- செவிலியர் எனும் தாய்
- HEPATOLOGY FOR NURSES
- Editorial
- Editorial Board
- Unknown etiology, thoughtful management, gratifying outcomes!
- Antiphospholipid Syndrome(APLS) in a man
- World Organ Donation Day Celebration at Kauvery Hospital, Hosur
- Approach to cardiac arrest
- Cardiac Tissue Viability Study
- Dietary management after Mitral Valve Replacement
- Clipping of a Cerebral Aneurysm at Nellai
- Care of Chronic Suppurative Otitis Media (CSOM)
- Pituitary Macroadenoma: A case report
- Superior mesenteric artery thrombosis
- Pharmacists: The Silent Heroes of the Health Care System
- Educative Image
- Poem
- A battle to win a baby
- 5S Sustenance Level-Up for Salem, Hosur and ECB Units
- Editorial
- Prevention of accidental removal of tube and drains: A case report
- Clinical Report Workshop: An initiative by the Clinical Governance Team
- Burr Hole – evacuation of right frontoparietal chronic Sub Dural Hematoma and left parietal SDH
- Balloon Mitral Valvotomy
- Intra Pulmonary Thrombolysis
- Nephrotic Syndrome: A case report
- Trans catheter Aortic Valve Implantation (TAVI)
- Tetralogy of Fallot (TOF): Echocardiography
- Patent Ductus Arteriosus (PDA)
- A souvenir to remember the Nurses day 2024: Celebration of the Guardian angel of Healthcare
- Flying Cherub
- காவேரியின் செவிலியராய்!
- காவேரியின் பசுமை புரட்சி
- Editorial
- Care of patient with Interstitial Lung Disease
- Ovarian Torsion: A case report and discussion
- Ovarian torsion: A case report
- Acute Respiratory Distress Syndrome: A case report
- Shadow reports of heart city’s valve diseases and clinic
- Innovative strategy, empowerment and amendment of guidelines to prevent IV complications
- Effectiveness of Nurse- fabricated innovative device (K-Brace) on prevention of phlebitis
- Organ Donor Hero!
- Understanding Thalassemia: A comprehensive overview
- Pressure Injuries—the sensitive indicator
- The sixth QOK Competition April 2024
- Editorial
- Infective Endocarditis (IE)
- A case report on Bull Gore Injury
- Free Flap for traumatic raw area: A case report
- The Story of the “First Cry”
- Case series on drug-induced anaphylactic shock
- Clinical therapeutics: The adrenergic system and related drugs
- Flying angel experience
- From QOK to KOACH: A journey of continuous improvement
- Poem – காவேரியின் நவீன ஐந்து எஸ்(5S) மாடல்
- Editorial
- Editorial Board
- Attempted Hanging: A case series
- Sub Arachnoid Hemorrhage due to Cerebral Aneurysm
- Acute Pulmonary Thromboembolism with Systemic Lupus Erythematous
- Down Syndrome with Severe Pulmonary Stenosis
- Bentalls Procedure
- Glycogen Storage Disease (GSD)
- Nursing Care of Patient with Penetrating Chest Injury (Left Chest Wall)
- A child with Acute Inflammatory Demyelinating Polyradiculoneuropathy (AIDP) – Guillian- Barre Syndrome (GBS)
- Periampulatory carcinoma (Whipple’s procedure)
- Assisted delivery with Forceps Extraction
- Thrilling Moment on a Successful transportation of the cadaver donor liver to its recipient
- Editorial
- Transcatheter Aortic Valve Implantation (TAVI)
- Cautery Burns: A Clinical Audit
- Automatic Implantable Cardioverter Defibrillator (AICD)
- Thymectomy
- Deep Vein Thrombosis: A case report
- Mesentric Neoplasm
- QT Syndrome
- Healthy diet, affordable for all – Fuel for the Future
- Personalizing 5-FU Treatment in Head and Neck Cancer: A TDM Pilot Study
- Rapid Review of CNE -Nursing Challenges in Coronary Artery Diseases
- Preconference Workshop on International patient safety Goals (IPSG)
- Editorial
- Cerebral malaria: Management with artesunate
- Hypertrophic cardiomyopathy: A case report
- Incredible challenges and outcomes: A case report
- Prevention of extravasation in Oncology unit
- Management of germ cell tumors: A review
- Monocytes: The mysterious cell on the CBC
- Efficacy of individualized use of a multisensory integrative environment on engagement: In children with sensory modulation disorder
- Dietary guidelines and food safety: For immuno-suppressed/compromised patients
- Nutritional management: For a patient with triple vessel disease
- இயன்முறை மருத்துவமும் மறுவாழ்வும்
- உயிர் காக்கும் தானம்
- மாற்றத்தை விதைக்கும் திறனாளிகள்
- EDITORIAL
- EDITORIAL BOARD
- CEREBRAL MALARIA: A CASE REPORT
- CEFEPIME – TAZOBACTUM – INDUCED FLUID – FILLED BLISTERS (BULLOUS LESIONS): A CASE REPORT
- BLOOD TRANSFUSION REACTIONS: AN OVERVIEW
- EFFECTIVENESS OF REHABILITATIVE APPROACH-BASED MANAGEMENT FOR CHILDREN WITH JAPANESE ENCEPHALITIS (JE)
- THE EFFECT OF NUTRITIONAL COMPOSITION ON THE GLYCEMIC INDEX AND GLYCEMIC LOAD VALUES
- KCHS PRATIDHI RISES TO THE CHALLENGE: A TRIUMPH AT THE NATIONAL QC CONVENTION
- JOURNAL SCAN FOR THE CLINICAL PHARMACIST
- காவேரித்தாய் – 4
- EDITORIAL
- INSTRUCTIONS TO AUTHORS
- AORTIC ANEURYSM WITH PARAVERTEBRAL COLLECTION
- DEEP VEIN THROMBOSIS AT RIGHT UPPER LIMB: A CASE REPORT
- ENTEROVIRUS ASSOCIATED MENINGOENCEPHALITIS: A CASE REPORT
- CONGENITAL HEART DISEASE (ASD – OS): A CASE REPORT
- FUNCTIONAL SHORT GUT SYNDROME: A CASE CAPSULE
- PERI-OPERATIVE CARE DURING CYTOREDUCTION SURGERY (CRS) WITH HYPERTHERMIC INTRAPERITONEAL CHEMOTHERAPY (HIPEC): KAUVERY EXPERIENCE.
- A CASE REPORT: MYELOPROLIFERATIVE NEOPLASMS (MPNS) WITH CABG
- OVERVIEW OF BREAST FEEDING: A REVIEW
- PITUITARY MACRO ADENOMA-TRANS-NASAL TRANS-SPHENOIDAL ENDOSCOPIC EXCISION
- JOURNAL SCAN: A CASE REVIEW OF IMMEDIATE CLINICAL SIGNIFICANCE, HARVESTED FROM MAJOR INTERNATIONAL JOURNAL
- தயக்கம் தவிர்
- Editorial
- Editorial Board
- Case Report: A success story of IVUS: In a PTCA
- Case Report on Ovarian Torsion
- Nursing care for a child with Thrombophlebitis
- Drug Induced Hypersensitivity to Salazopyrin: A case report
- The Phytonutrients — 365.25 Days/52 Weeks of Phytonutrients help’s to develop our shape
- Role of diet in coronary artery disease
- 5S Cross Unit Assessment 2024
- என்னுள் 5S
- பெண்ணே பெருமை கொள்
- Journal scan
- Editorial
- Instructions for Authors
- Ventricular Septal Defect (VSD): Echocardiography
- Steven Johnson Syndrome: A case report
- A case review on Capsule Endoscopy
- Savoir Faire: A management for mass causality
- Case report on Gouty Arthritis
- Management of a road traffic accident victim with severe head injury, and aspiration: A case report
- A case report on inhalation of chlorine gas
- ST segment elevation during Treadmill exercise test in a patient without prior Myocardial Infarction
- History and evolution of Corporate and Clinical governance
- The need of Human Papilloma Virus (HPV) vaccine: A review
- Futuristic face of resuscitative Centhaquine for hypovolemic shock: A Review
- Boost your health in this summer
- A data-driven approach to patient care: Kauvery Hospital pioneers six sigma in healthcare
- Fascinating experience as a flying angel
- விளையாட்டுத் திடல்
- எங்கள் காவேரி
- Editorial
- Triple Bypass: A case report
- Boerhaave Syndrome with Mediastinitis
- Right sided Infective Endocarditis
- Marburg virus disease: A systematic review
- Advances in cardiac amyloidosis treatment: A review on Tafamidis
- Role of salt in human health
- Learning by playing: 5S makes school a breeze
- நம் காவேரி
- செவிலிய தேவதைகள்
- Editorial
- A case report: Melioidosis
- Type IV-A Choledochal cyst
- Nursing care of the patient with right lower lung foreign body and Bronchiectasis: Treated surgically with lobectomy
- Care of patient with OPC poisoning
- The Rosai-Dorfman disease presents with extranodal involvement and diagnostic challenges
- Open stab injury by a Bull
- In-house Continuing Nursing Education (CNE) on mastering the complexities of critical illnesses at Kauvery Hospital, Tennur, 2024
- Effectiveness of trigger point release and ultrasound therapy on Trapezitis
- Kauvery Hospital Trichy region’s journey towards 5S model hospital recognition
- A Triumphant journey to 5S sustenance level I
- Poem – இரத்த தானம்
- Poem – ஆசிரியர் தின நல்வாழ்த்துக்கள்
- Poem – ஆசையான ஆசான்
- Poem – அவள் ஓர் அறிவியல்
- Editorial
- A Case of Burns
- A Case of Diabetic ketoacidosis
- A Case of Osteoarthritis
- A Case of Stroke
- A Case of Acute Myeloid Leukemia
- Case Study on Blunt Injury induced AKI
- Integration of Recent Technology in the Operation Theatre Enhancing Patient Outcomes
- Nursing Care of Patient with Congenital Acyanotic Heart Disease for Diagnostic Cardiac Catheterization Done
- May Thurner Syndrome Vascular Condition of Postpartum
- Myocardial Infarction in a Young Patient
- Nursing Care of Patient with Dextrocardia
- Case Study on Left Gangliocapsular Intracranial Hemorrhage with Cranioplasty
- Silent Leaks, Loud Alarms: Nursing Vigilance in a Complex Neurosurgical Recovery
- From Crisis to Care: The Challenge of Managing Myasthenia Gravis in an Elderly Patient with Multiple Comorbidities
- Dual Intervention Success: Managing Bradycardia and Heart Failure with TPI and CRT – D
- A Case Report On Mastitis
- Our Nurses-Our Future: Investing in Nurses Empowerment for Enhancing the Nursing Workforce
- Empowering Patients in Patient Care – Prevention of Medication Error
- Case Publication Report: Malignant Ascites and Pleural Effusion in Stage 4 Ovarian Carcinoma – An Elaborated Case
- Case Study: Management of Thoracic Endovascular Aneurysm Repair (TEVAR) in an 82-Year-Old Male with Type B Aortic Dissection
- Case Study: A Rare Presentation of Type A Wellens Syndrome in a 45-Year-Old Male with Type 2 Diabetes Mellitus and Systemic Hypertension.
- A case report on Endometriosis
- Molar Pregnancy: A case report
- Review article of immune checkpoint inhibitors in cancer patients
- Case Study: Management of Kawasaki Disease in a Pediatric Patient
- Empowering Nurses through Hands-on BLS Training: A Large-Scale RNRM CNE Renewal Initiative by Kauvery Hospital, Trichy
- Editorial
- Empowering Clinical Participation Nurses Through Academic and Research participation
- From policy to practice: Transforming nurse competence through a restraint management recall program
- Peritoneal equilibration test in our CAPD patients: A retrospective analysis
- Perinatal asphyxia with hypoxic-ischemic encephalopathy stage I in a late preterm neonate: A case report
- A case of vibrio cholera
- Emergency nursing management of a patient with acute aortic intramural hematoma
- Case report on testicular cancer
- Case study on abdominal tuberculosis
- Monoclonal IgG kappa (IgGk) associated crescentic glomerulonephritis: A case of PGNMID in disguise
- Critical management of upper gastrointestinal bleed with septic shock in an elderly patient
- My experience in a renal transplant ICU
- Pulmonary vein stenosis
- A case report: Rheumatic heart disease and congestive heart failure in antenatal mother
- Case of corrosive poisoning with pneumonia
- A case report on Stevens Johnson Syndrome
- A case report on Sub Arachnoid Hemorrhage (SAH)
- Autosomal Recessive Polycystic Kidney (ARPKD) with cavernous transformation of portal vein
- A case report on open heart valve replacement
- Editorial
- The impact of home-based physiotherapy on functional capacity and quality of life in patients with severe heart failure
- Liver transplantation
- The Invisible Man – Androgen Insensitivity Syndrome: Disorders of sexual development
- Pericardial effusion
- A case report and discussion: Burns
- Challenges of polypharmacy in a geriatric patient with neurological disorder
- A case report on Bullous Pemphigoid (BP)
- Herpes Zoster Encephalitis: Diagnostic and Clinical Insights
- A Case Report & Review on Sternal Osteomyelitis
- Continuing Nursing Education on Prevention of Hospital Acquired Pressure Injury
- Prevention of Hematoma and Thrombus After CAG /PTCA
- Case Report: Neonatal Hirschsprung Disease
- Current Treatment, Challenges, and Research Updates in Sexually Transmitted Infections: A Detailed Review
- Lipoinjection for fat deficiency in right cheek
- LA Myxoma
- Nursing care of Sturge – Weber Syndrome (SWS), referred for Digital Subtraction Angiography (DSA)
- Nursing care of patient with Sick Sinus Syndrome
- Post-Partum Acute Kidney Injury
- Service Uniqueness and Management Outcomes (SUMO) in Healthcare Services
- Poem – அம்மா!!!
- Editorial
- When Banding Breaks, New Paths Awaken: The BRTO Revelation
- Smile Therapy
- Multidisciplinary approach to Thermal Burns
- Deep Brain Stimulation for Parkinson’s disease: A case report
- Zieve’s Syndrome: A review
- Acute Pulmonary Thromboembolism
- MPI scan guided revascularization in acute anterior wall Myocardial Infarction
- Ketogenic diet for Epilepsy: A case report and review
- Dietary management: Carcinoma in left buccal mucosa
- Malignant Middle Cerebral Artery (MCA) infarct and surgical decompression: Pre-op and post-op CT brain findings
- Cleistanthus collinus (Oduvanthalai poisoning): A case report
- My Experience as a Flying Angel
- In-house Continuing Nursing Education (CNE) on “Rapid Response Mastery
- Kauvery Hospital Salem’s Journey of 1st Ever Model Hospital
- மனமும் வெற்றியின் ரகசியமும்
- Editorial
- Against all odds: A road accident survivor’s journey to healing at Kauvery Hospital
- Clinical Case Report: Managing Hansen’s Disease in a 20-Years young girl
- Bilateral Internal Thoracic Artery Grafting for CABG
- Intra Pulmonary Thrombolysis
- A Case Report on Methotrexate-Induced Pancytopenia
- An Adult with an Atrial Septal Defect Presenting with a Brain Abscess
- Typhoid, a Prospective Observational Study
- Vancomycin – Therapeutic Drug Monitoring
- Cardiac’s Myxoma
- Mitral valve replacement
- Harmful effects of preservatives (Class 1) on Food Items
- In house Continuing Nursing Education (CNE) on “Shaping Excellence in Critical Care Nursing.” At Kauvery hospital, Cantonment.
- Poem – செவிலியர்
- Poem – ஒருபோதும் கேட்காதீர்கள்: “உனக்கு என்ன வேண்டும் என்று”
- Editorial
- A case report on Carbuncle
- Reverse Shoulder Arthroplasty: A case report
- A case report on severe dental caries with advanced lesions
- Supra ventricular Tachycardia: A case report
- A case of pernicious anaemia due to vitamin B12 deficiency
- A Journey of Miracles: Life Beyond the Deadly Trials for My Father
- A Victory day for CNE
- A Sapient Voyage – QCFI
- Tracheostomy: An overview
- முன்கூட்டியே கண்டறிவோம் புற்றுநோயை
- Editorial
- Emergency CABG for young female patient with critical coronary artery disease
- Meningomyelocoele: A case report and discussion
- Case study on Multiple Cranial Nerve Palsy and Necrotizing Pneumonia: The physiotherapy management
- Role of Physiotherapy in ACL Rehabilitation: A case report
- ASD Device Closure: Case report and discussion
- In-House-Continuing Nursing Education (CNE) on “Effective Nursing Strategies for Renal Transplantation” at Kauvery Hospital, Tennur
- காவேரியின் வாக்ஹோலிக் நடைபயிற்சி
- புத்தாண்டு
- Editorial
- Artificial Intelligence in Nursing: Enhancing Care and Reducing Burnout
- Report on comprehensive wound care workshop—elevating nursing excellence at Kauvery Hospital
- Cerebellopontine angle tumor
- Patient acuity score: Staffing plan
- Acute Respiratory Distress Syndrome
- Coronary Artery Disease and Carotid Stenosis: A dual threat
- Early-onset diabetic foot ulcers in CKD
- Nursing case study report: Reconstructive surgery for congenital TMJ ankylosis
- Care of severe ARDS and H1N1 Positive
- Whipple Procedure: A case report
- A milestone to remember in my career
- Poem – காதல்
- Poem – ஆரோக்கிய வாழ்வு – 2
- Editorial
- Management of Myelodysplastic Syndrome (MDS) with Probable Fungal Pneumonia
- Thrombotic Microangiopathy and Renal Cortical Necrosis in a Postpartum Patient: A rare and complex presentation
- Rising Star in Health care
- Systemic Lupus Erythematosus: A case report and discussion
- Effectiveness of Cardiopulmonary Resuscitation( CPR) and its Outcome
- Guillain-Barre syndrome
- Radiation-free ERCP in pregnancy
- Utilization of injection Sovateltide for acute ischemic stroke
- A case of severe malaria complicated by concurrent H 3 N 2 influenza infection: Diagnostic and therapeutic challenges
- Pulmonary Function Test Concepts
- Rapid Review of CNE – Enhancing Nursing Practice in Arrhythmia Management: Evidence Based Strategies
- நூறைக் கடந்த காவேரியின் மருத்துவ இதழ்(ஜர்னல்)
- பெண் என்பவள்
- வியக்கத்தகும் அதிசயமே! கண்டு வியக்கிறேன்
- Editorial
- Early Rescue PCI in Failed Thrombolysis in STEMI
- Internal Jugular Vein Thrombosis: A Case Report and Discussion
- The Beat of Compassion: A Clinical Presentation of Nursing Excellence
- Acute Necrotizing Pancreatitis: Challenges in Management and Recovery
- “From Struggle to Breathe to Freedom to Live”: The Miracle of Pulmonary Thromboendarterectomy
- Waugh Syndrome (Ileocolic Intussusception +Malrotation): A Case Report and Discussion
- Corrosive Poisoning: A Case Report
- Multiple Intracranial Aneurysms: A Case Report and Discussion
- Steroid-Dependent Nephrotic Syndrome in Pediatric Patients: Pharmacologic and Preventive Management
- Shared Decision-Making should be an Integral Part of Physiotherapy Practice: A Case Study on Total Knee Replacement
- NICU Graduate Day: “Saving the Unsavable” by Trusted Quality Care
- Balancing Technology and Patient Safety: Insights from the Workshop
- Impact of Nurse Leadership on Patient Outcomes
- பெற்றெடுக்காத அன்னை
- மனம் – ஒரு மாயை!
- Editorial
- Idiopathic Parkinson’s Disease
- A case report on Guillain–Barré Syndrome
- A case of Iatrogenic Mediastinitis
- A case of puerperal sepsis due to ESBL E. coli with multi-organ involvement: A clinical challenge
- Critical management of severe obstructive cholangitis with septic shock in an elderly patient with cardiac and renal comorbidities
- Acute cholecystitis after cardiovascular surgery (CABG)
- Comprehensive management of diabetic cellulitis in hand and its outcome
- A case of successful kidney transplantation after a long-term maintenance in haemodialysis
- “Mystery of Blue boy” Methemoglobinemia poisoning: Challenging in identification and treatment
- A case report on ovarian cyst torsion: Emergency procedure
- A case report on status epilepticus
- A new lease on life: Successful discharge after brain tumor
- The road to recovery: A case study on liver transplant success
- Systemic Lupus Erythematosus: A case report and discussion
- Carpal tunnel release surgery: A nursing case study on post-operative care
- Evidence-based nursing practice: A case study on Zadek’s procedure for ingrown toenail”
- Against the Odds: Impella-supported revival in an octogenarian with cardiogenic shock (stage E) and advanced coronary artery disease
- A structured approach for patient safety and experience: Enhancing traditional nursing practices with new dimension
- Nursing care of patient with penetrating left chest pain
- Through the crack of a blast, light of care found its way—Multisite Blast injury in a Farmer from Improvised Explosive Device: A Nursing Perspective
- Pulmonary Tuberculosis: A case study and clinical perspectives
- Secondary Postpartum Hemorrhage
- Care of patient with spondylodiscitis
- Nursing care of patient with cauda equina syndrome
- Editorial
- A case of spinal tuberculosis with acute spastic paraplegia managed with medical therapy and surgical fixation: A comprehensive clinical and nursing perspective
- Antiphospholipid antibody syndrome presenting as pulmonary thromboembolism and diffuse alveolar hemorrhage in a young female
- Ethical and clinical management of a jehovah’s witness patient undergoing deceased donor renal transplantation
- Effective management of type II endoleak post EVAR: A multidisciplinary approach by nurses
- Case Study: “Successful TAVI procedure for severe aortic stenosis, a patient’s journey”
- CRRT: More than renal replacement, a case study in multiple organ support
- Clinical practice guidelines on peripheral IV therapy practices
- Gulliain Barre Syndrome: A case report
- Case Report: Multiple sclerosis in a 28-year-young female
- Comparative case study report: Paraquat poisoning with multiorgan dysfunction
- Naegleria fowleri (Brain-Eating Amoeba): A comparative epidemiological and pathophysiological review—Global, Indian, and Kerala perspectives (2025)
- Patient safety colloquium 2025: “Safe care for every patient, every time”
- A lifesaving miracle: Bone marrow transplant gives six-month old baby a new lease of life
- The Healing of broken wings: A case of paediatric firecracker injuries
- Ureterovaginal Fistula Following Hysterectomy – A Clinical and Nursing Management Case Report
- Case Study: Adult-Onset Henoch–Schönlein Purpura (HSP)
- Desidustat: Role in management of anemia of chronic kidney disease (CKD)
- Comprehensive clinical management of an extensive lumbo-sacral wound with multiple sinuses in an elderly male
- Autoimmune encephalitis with anti-LGI1-antibody: A case report
- Case presentation on Total Knee Replacement
- In-House-Continuing Nursing Education (CNE): “Nursing Perspectives in Oncology”, Kauvery Hospital, Tennur
- Editorial
- Abdominal aortic aneurysm repair
- A case of ovarian cyst with partial torsion in an adolescent girl
- Clinical presentation and nursing care of a patient with acute ischemic stroke
- Nursing care of young patient with aortic valve replacement for bicuspid aortic valve and aortic valve stenosis
- Young patient care of abdominal aortic aneurysm repair
- Nursing management of multiple diagnoses and thrombectomy procedure
- Bypass to the future: A CABG success story
- A case report on pharyngeal fistula
- Chandipura virus: an overview
- Nursing case study: Management of right Common Iliac Artery (CIA) stenosis with Chronic Total Occlusion (CTO)
- Case study on peripheral arterial disease and its complications: Chronic limb threatening ischemia
- Foreign body removal: A case report
- The role of breast milk in enhancing nutritional and immunological properties
- A case report on sub dural hematoma in a patient on dual antiplatelet therapy
- Highly concentration electrolytes: A silent risk every nurse must recognize
- Transverse limb defect: A case report
- A quick review on the management of myasthenia gravis during pregnancy
- Youngest pediatric bone marrow transplant: Thalassemia major donor—thalassemia major marked sibling, bone marrow transplantation
- MOG antibody-associated optic neuritis: A case report
- Stuttering cerebrovascular accident and a rare intervention: A case report
- Thoracotomy with pleuropericardial window
- Enhancing healthcare leadership and sustainable team building through the OODA loop: insights from Kauvery hospital
- Clinical spectrum and management outcomes of GDM and GHTN: A case series from a tertiary care center
- The future of insulin: Innovations, AI, and the evolving role of pharmacists in diabetes care
- Editorial
- A bond beyond words—a beautiful truth when one twin can be the sibling’s life saver
- Acute limb ischemia in a young female with systemic lupus erythematosus
- A case report on laparoscopic retrieval of a massive gastric trichobezoar causing partial gastric outlet obstruction in a paediatric patient
- A complex neonatal presentation requiring Ladd’s procedure: A case report
- Effect of diet and lifestyle modifications on non-pharmacological HbA1c reduction in a new onset diabetes pre-renal transplant donors
- Pioneering the future: The first step in robotic surgery
- Neuromyelitis Optica
- Sleeve gastrectomy
- Multidisciplinary nursing approach to D6 vertebral collapse secondary to multiple myeloma
- Probable hereditary haemolytic anaemia with multisystem complications: A one-month clinical journey
- Type B Thoracoabdominal Aortic Dissection (TBAD) with fusiform aneurysm of infra-renal abdominal aorta
- Acute viral hepatitis
- Functional outcome of MOD QUAD procedure in a child with right sided Erb’s Palsy (Right Upper Limb)
- Paediatric hemangiomas of right lower eyelid excision: A case study from a nursing perspective
- Nutriotic table
- RTA with polytrauma injury
- Nutrition week celebration at Maa Kauvery Hospital
- Continuing nursing education on nurse’s role in infection prevention and control, in critical care unit
- Two type data analysis: Qualitative data vs Quantitative data
- Journal scan
- Editorial
- Dual kidney transplantation: An Emerging Strategy to Expand the Donor Pool
- A case of critical management of ILD
- A case of bilateral Congenital Talipes Equinovarus (CTEV)
- A case of gastric volvulus
- Penetrating stab injury to the neck (Zone II) with left subclavian artery injury and massive haemothorax
- Workshop on clinical audit and quality improvement projects: Strengthening nursing practice
- A case report on surgical retrieval of fragmented intravenous cannula tip from the cephalic vein
- The left sided mystery: Situs inversus totalis
- A challenging case of prolonged fever with acute kidney injury and multifocal neurological involvement
- Neglected right testicular tumor presenting as metastatic pleural empyema
- Successful pregnancy outcome in a primigravida with sick sinus syndrome on permanent pacemaker and previous cerebrovascular accident
- Exclusion of infra renal abdominal aneurysm with aorta bi-iliac bypass
- Cerebral fat embolism in a young polytrauma patient: A case report
- Dual surgical intervention in cardiac disease: A case study on CABG with AVR
- Successful management of refractory supraventricular tachycardia in a 4-month-old infant
- First successful neonatal percutaneous cardiac intervention done in Maa Kauvery & Heart city, Trichy
- Neuro Nursing – Quality Markers
- Drug of the month: Nitric oxide & nitrous oxide– Same family, different stories
- Multiple tablet overdose with multi-organ involvement and critical illness neuropathy: A case report from Kauvery Hospital, Trichy
- Amyotrophic Lateral Sclerosis (ALS): A case report
- Weight management for cancer patients
- Journal Scan
- Editorial
- A new milestone in spine care: spine endoscopy experience at kauvery hospital, trichy (Cantonment) – 2025
- Ethylene Glycol Poisoning Using Ethanol Therapy
- Robotic total knee replacement: First of its kind experience at Kauvery hospital, cantonment, Trichy – 2025
- Enhancing the safety of hospitalization by reducing patient falls
- Alcohol septal ablation for severe hypertrophic obstructive cardiomyopathy during pregnancy: Rare presentation
- C6 vertebral fracture secondary to tuberculous spondylodiscitis in a patient with chronic kidney disease
- Idiopathic intracranial hypertension presenting as recurrent cerebrospinal fluid rhinorrhea
- Critical pertussis
- Thinner Ingestion (Hydrocarbon Poisoning)
- Anaesthetic management of Oberlin Nerve Transfer without NMN-Depolarising muscle relaxants
- Tb meningitis–associated CNS vasculitis and stroke in HIV and diabetes: Highlighting multidisciplinary management
- From passive to active: Clinical reasoning in acute care chest physiotherapy
- Revision detethering surgery in a child with tethered cord syndrome associated with split cord malformation type II
- Thymoma (Anterior Mediastinal Mass)
- Laparoscopic bilateral etep mesh repair of bilateral recurrent right giant inguinoscrotal hernia + resection anastomosis of small intestine
- Management of post-renal biopsy pseudoaneurysm and rapidly progressive renal failure in a patient with IgA nephropathy
- Poly trauma patient care: A nursing approach
- Lenacapavir and the persistent challenge of HIV eradication: Biological barriers, therapeutic innovation, and future research directions
- Medivault: standardized storage system for medication safety in inpatient wards – A kaizen-based quality improvement initiative
- The Gateway to Quality Care: Evidence-Based Initial Patient Assessment
- Ultra-rapid thrombolysis in 19 minutes: Breaking conventional door-to-needle benchmarks
- Distinguished Alumni: A journey rooted in Kauvery
- Journal Scan: A review of images in clinical medicine of immediate clinical significance, harvested from major international journals
- Editorial
- Prolonged cardiac arrest during primary PCI in acute anterior wall myocardial infarction
- ASD with Partial Anomalous Pulmonary Venous Connection (PAPVC)
- Quality Improvement to improve patient safety and satisfaction: Effective whiteboard communication tool.
- A comparative study on dysphagia management & optimization of nutritional status among acute stroke patients
- Clinical case study on management of glycemic variability in gestational diabetes mellitus, diet as a primary therapeutic tool
- A rare case of multiple large bowel perforations following air blast injury through the rectum
- Carcinoma of the lower esophagus managed by video-assisted thoracoscopic esophagectomy
- A tale of two twins: A detailed case presentation of preterm neonates with divergent clinical courses
- From fragile lines to fortified care: A nursing-led revolution in iv dwell time at Kauvery hospitals
- Navigating the two-stage elephant trunk procedure (CET): A nursing case study on postoperative vigilance and multidisciplinary care
- The 3C initiative: weaving a stronger fabric of care through monthly connection at Kauvery hospital Vadapalani
- ACL reconstruction rehabilitation in an adolescent athlete: A case report
- Myasthenia gravis
- Comprehensive care for Preterm & Low birth weight babies
- National Cardiac Life Support (NCLS) workshop at Kauvery Hospital
- Strengthening institutional commitment to patient safety: Report on a structured training workshop at Kauvery hospital
- Dietary management for the RTA with polytrauma
- Successful staged Percutaneous Coronary Intervention (PCI) in a patient with cardiogenic shock and triple vessel disease
- Early infantile developmental epileptic encephalopathy
- Peutz Jeghers Syndrome
- Emergency nursing management of a patient with? Meningitis and spinal abscess
- Report on continuing nursing education (CNE) program
- Dual lipid-lowering therapy: Maximizing IDL-C reduction through complementary mechanisms
- Calvarial Osteomyelitis & CSF Leak Management Post Pituitary Surgery
- Journal Scan
- என் செவிலிய பணி
- Editorial
- Diffuse axonal injury grade III
- Cerebral venous thrombosis
- Arrhythmogenic right ventricular cardiomyopathy presenting with sustained ventricular tachycardia in an adolescent male: A case report
- Imaging the Invisible: Paediatric Venous Malformation of the Ankle with DSA Confirmation
- Clinical pharmacy perspective: Can antiplatelet therapy be used for preventing thrombus formation in the left atrium and apical clot?
- Left carotid endarterectomy with coronary artery bypass grafting (CABG)
- Left pulmonary artery stent restenosis in a patient with repaired tetralogy of fallot
- Preventing the preventable: Clinical pharmacy role in managing severe hypoglycemia in an elderly patient on glimepride
- Severe aortic regurgitation with ascending aortic dilatation in a young adult with bicuspid aortic valve
- A Case Study on Surgical Repair of Subclavian Artery Aneurysm.
- Thoracoabdominal and suprarenal Abdominal Aortic Aneurysm: Surgical and Critical Care Management
- Heart to heart illness for mother and daughter
- Clinical Pharmacy of Torsemide: Toxicity and Safety Considerations in Elderly Patients – With an Embedded Case Report
- Tetanus with Autonomic Dysfunction in an Elderly Diabetic Patient with Restrictive Cardiomyopathy and Implanted Pacemaker: A Fatal Outcome
- Case study on MODY (Maturity onset diabetes of the young) – Taper down of the sulfonylurea doses
- Effectiveness of cryo-compression therapy in preoperative and postoperative orthopaedic patients: A case study
- Emergency laparotomy in a patient with bleeding duodenal ulcer
- Successful management of severe pre -eclampsia complicated by massive proteinuria and fetal growth restriction in a primigravida
- Blunt thoracoabdominal trauma with rib fractures and muscle laceration: A case report
- Flash fire injuries: A preventable catastrophe
- The River Within: A Journey of Life and Renal Care
- Evidence based management of cervical insufficiency
- Treacher Collin’s Syndrome
- Effective nursing triage and stroke code activation in acute hypertensive intracerebral haemorrhage presenting with sudden loss of consciousness
- Euglycemic Diabetic Ketoacidosis: A Diagnostic Challenge in Clinical Practice
- IV – Nurse Day Celebration-2026
- Nutritional management in deceased donor liver transplantation: A case study of decompensated chronic liver disease with hepatocellular carcinoma
- The body’s sudden shutdown: Vasovagal syncope
- Weaning from Ryle’s Tube Feeding to Oral Diabetes Diet in a patient with Diabetes, and with Respiratory Compromise
- Successful management of traumatic lung injury using veno-venous ECMO in a young RTA patient
- Cne certification program on prevention and management of venous thromboembolism (VTE): Risk assessment, early recognition, prevention, and evidence-based management
- Nurses day celebration 2026: A week of recognition, learning, and empowerment
- Electrical shock and its management
- Diagnostic approach to connective tissue disorder: Spotlight on adult-onset still’s disease
- Gastric varices in chronic liver disease managed with parto procedure
- Pararenal abdominal aortic aneurysm – preparation is the key to successful repair
- Journal Scan: A review of images in clinical medicine of immediate clinical significance, harvested from major international journals
- தாயும் சேயும்
